|
|
|
| Charcateristic Dentofacial Features In Apert's Syndrome (acrocephalosyndactyly) - A Case Report |
Sumati Bhalla 1 , Manpreet Kalra 2 , Parampreet K. Pannu 3
1 Sr Lect., Dept. of Public Health Dentistry - Dr. Harvansh Singh Judge Institute of Dental Sciences & Hospital, Chandigarh U.T.- 160014, INDIA
2 Reader, Department of Oral Pathology - S G T Dental College, Gurgaon
3 Professor and HOD, Dept. of Pedodontics - Gian Sagar Dental College and Hospital, Patiala
|
| Address For Correspondence |
Dr. Sumati Bhalla, Senior Lecturer
Department of Public Health Dentistry
Dr. Harvansh Singh Judge Institute of Dental
Sciences & Hospital, Panjab University, Sector 25
Chandigarh U.T.- 160014, INDIA |
| Abstract |
| Apert’s syndrome is a rare congenital anomaly characterized by acrocephaly, syndactyly, midface hypoplasia, pharyngeal attenuation, ocular manifestations and abnormalities of other organs. It has characteristic features in the orofacial region like early craniosynostosis of coronal suture, cranial base and an agenesis of the sagittal suture. These characteristics predispose the patients to maxillary transverse and sagittal hypoplasia, pseudo cleft palate as well as prominent skeletal and dental anterior open bite. In this case report, the features of Apert syndrome, particularly in relation to the orofacial region, are discussed with an emphasis on the need for multidisciplinary care in such patients. |
|
| Keywords |
| Apert’s syndrome, acrocephalosyndactyly, pseudo cleft palate, mid facial hypoplasia |
|
| Full Text |
Introduction
We ought not to set them aside with idle thoughts or idle words about “curiosities’ or “chances”. Not one of them is without meaning; not one that might not become the beginning of excellent knowledge, if only we could answer the question- why is it rare, or being rare, why did it in this instance happen? James Paget, 1882[1]
Over the past several decades tremendous advances have been made in the prevention and treatment of developmental anomalies. This metamorphosis in our conceptualization of developmental malformations has led to an improved ability to handle and prevent them. Despite such improvements, developmental malformations remain a significant cause of morbidity worldwide. Even when the mode of inheritance is well established, some conditions continue to exhibit a large number of sporadic occurrences, which makes their eradication virtually impossible. As such, it is incumbent on us to learn us much as possible about these conditions. In this way, we can become better clinicians and impart better care to those who so desperately need it.[2]
Acrocephalosyndactyly is a rare developmental deformity characteristically affecting the head, hands and feet. Eugene Apert[3] in 1906 reported nine cases and since then his name has been associated with acrocephalosyndactyly. Apert described a triad of craniosynostosis, syndactyly and maxillary hypoplasia. It is known to be inherited in an autosomal dominant fashion, but most cases are sporadic. The sporadic cases are postulated to be associated with advanced paternal age. The incidence of Apert syndrome is approximately one in 50,000 births. Some investigators state that 4.5 percent of all craniosynostosis represent Apert syndrome.[4]
Apert's syndrome has been rarely reported from India.[5] With the rare exceptions, Apert's syndrome in all reported cases has been caused by recurrent missence mutations of the fibroblast growth factor receptor 2 gene involving 2 adjacent aminoacids.[6] Apert's syndrome is thought to occur as a result of androgen end organ hyper-response affecting the epiphyses and sebaceous glands.
This results in early epiphyseal fusion resulting in short stature, short and fused digits and acrocephaly. The clinical features are characterized by early fusion of skull bones, mainly coronal sometimes lambdoid, midface regression and webbed digits (syndactyly). Syndactyly always involves fusion of the soft tissues of the first, middle and ring fingers. Thumb may be fused with the rest or may be free.[7]
The middle third of the face is retruded and hypoplastic. The calvarial coronal synostosis and the saggital and metopic suture agenesis coupled with early synostosis of the cranial base results in a hypoplastic midfcae and a vertically accentuated craniofacial complex. Proptosis, downslanting laterla canthi are seen and often there is some degree of optical hypertelorism. The ears are often low set. The nose can have a parror beek shape with a depressed nasal bridge. The maxilla is hypoplastic in all three dimensions and is retropositioned. The palate is high arched and narrow due to poor aeration of maxillary antra. There are bulbous lateral palatal swellings (containing hyaluronic acid) which make the central furrow of the palate very prominent and difficult to cleanse. Pseudo cleft palate along with an anteriorly tipped palatal plane is very common. The maxillary arch is V shaped and there is severe dental crowding. The maxilla slants down posteriorly as a result there is anterior open bite. Impactions, severe crowding of developing teeth within the alveolus, delayed eruption, thick gingiva and sometimes supernumerary or congenitally missing teeth are the hallmarks of the maxillary dental development in Apert’s syndrome patients. In the mandible these findings are less pronounced. The lips are characterized by the crossbow shape of the upper li or the trpezoidal shape of both the lips. The lips range from non competent to competent depending upon their ability to form seal.[8]
Patients affected with acrocephalosyndactyly usually have a norma lifespan. Therefore it is imporatnt to remember that facial defects which develop qquite early can lead to various types of social maladjustments. Surgical intervention in such cases is for morphological, functional and psychological benefits. Paul Tessier[9] has presented a comprehensive system of craniofacial surgery for the definitive and radical correction of deformities in Apert’s syndrome.[10]
Treatment involves multidisciplinary teamwork including Craniofacial surgeon, Neurosurgeon, Pediatrician, Speech pathologist, and an Orthodontist. Correction of hypertelorism can be undertaken by a facial advancement operation. These children invariably need speech therapy after the surgical correction of abnormalities is done.[11]
Clinical Synopsis
A twelve year old female patient had presented to the Dental OPD with a chief complaint of pain in the lower right back too since few days. The patient presented with unusual craniofacial and dental features, which prompted a further detailed analysis of the case. On questioning the parents it was found that the patient was known case of Apert’s syndrome.
This child Sheeshna was the second in the family, born to non-consanguinous parents after a normal labour. The mother's age was 28 years and the father was 35 years old. The patient was the product of a full term uneventful pregnancy with no known exposure to infection, drugs or irradiation. No similar malformations were known in either parent's family. Both parents and their other children were examined and found to be normal clinically.
The facial appearance was peculiar due to lateral displacement of both medial canthi and associated epicanthic folds, with prominent downthrust proptopic eyes and a flat nasal bridge. There was a flexion deformity of the elbows and knees with symmetrical deformity of both hands, which were short and stubby, and showed complete syndactyly of all the fingers with a short displaced thumb and a synonychia of the index, middle and ring fingers. The palmar aspect was spoon shaped. The feet showed a varus deformity with syndactyly of all toes. The other systems revealed no abnormality.
Extraoral examination revealed that she had an abnormal facies with acrocephaly, brachycephaly, flat occiput and a high prominent forehead. A mild facial asymmetry was present in this case. There appeared to be facial flattening with maxillary hypoplasia and anterior open bite. “Crossbow” shape of the upper lip and a protruding lower lip were noticed. The lips were potentially competent. Ears were low set and down sliding of the palpebral fissures were present with shallow orbits and exopthalmos (strabismus). Nasal bridge was depressed (Figures 1, 2).
 | Fig 1. Extraoral Frontal View
 |
 | Fig 2. Extraoral Profile View
|
Typical dental and skeletal findings of Apert’s syndrome (Figures 3, 4, 5) were observed on intraoral examination. The palate was high arched with a pseudo cleft in its posterior third. The patient had set of permanent teeth. There was severe maxillary crowding, anterior open bite, ectopic eruption and poor oral hygiene. Her face showed a dolicocephalic pattern. The over bite was - 4 mm and the overjet was - 1 mm due to forwardly placed lower anteriors. The maxillary dental arch was V shaper where as the mandibular arch was wider. Since the patient had reported with the chief complaint of pain in the lower right back tooth an IOPA radiograph was taken and which suggested deep caries involving the pulp. The tooth in question (46) was tender on percussion. Oral prophylaxis was done and Root canal treatment was performed on 46 and then it was permanently restored with amalgam post obturation.
 | Fig 3. Intraoral Front View
|
 | Fig 4. Maxillary Arch
|
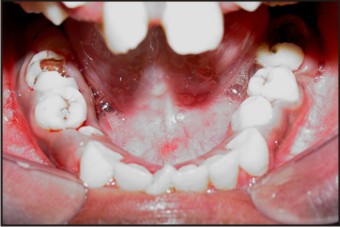 | Fig 5. Mandibular Arch
|
Discussion
A deformity of the osseous system is the most conspicuous feature of the syn-drome of acrocephalosyndactyly. It is recognisable at birth and characteristically affects the head and extremities.[12] The major manifestations include premature closure of the cranial sutures, and syndactyly affecting the hands and feet.
In most of the reported cases, the age incidence varies from 4 months to 9 years although the oldest patient reported has been 27 years of age.[13] In our case however, these deformities were noticed by the parents since birth.
The infant Apert skull is characterized by premature fusion of the coronal sutures and by a wide calvarial midline defect that starts at the glabella and ends at the posterior fontanelle. Many of those affected also have agenesis of the corpus collosum, progressive hydrocephalus, and hippocampal abnormalities[14]. Early surgical intervention to correct the craniosynostosis is crucial in order to realize the highest chances for normal development. The ocular orbits are shallow and the accompanying exophthalmia may lead to blindness. The ocular manifestations, hypertelorism and exophthalmia, seem to be present in most of the case reports and in this particular case as well.
Due to closure of the coronal sutures, the calvarium is lengthened vertically and shortened in the antero-posterior dimension, resulting in a flattened occiput and a prominent frontal area. There is hypertelorism with an antimongoloid slant and bulging of the eyes secondary to the shallow orbits. Facial dysostosis consisting of hypoplasia of the maxillae, a prominent mandible, high arched narrow palate, crowded teeth, an open mouth secondary to nasal obstruction, and occasionally a cleft palate are pre-sent.
The syndactyly is marked and resembles a `mitten hand' or `sock foot'. There is usually a complete fusion of the distal soft tissues, and occasionally of the bones. The thumbs and big toes may or may not be involved in the fusion. Other less frequent skeletal anomalies may be present.
Some mental impairment is present in almost every case, but its true incidence is not known. Most cases show no visceral abnormality. In this case no mental abnormality was found in the patient as her IQ score fell well within the normal range.
The oral cavity of Apert patients is also characteristic. The findings include a reduction in the size of the maxilla, particularly in the anterioposterior direction. This reduction may result in tooth crowding. Pseudo cleft palate or bifid uvula is found in approximately 75 percent of those affected. Dental anomalies such as impacted teeth, delayed eruption, ectopic eruption, supernumerary teeth, and thick gingiva are also common.[4] Most of these findings were observed in this patient.
The etiological factors prominent for this condition remain controversial. Previous reports favor the idea of a dominant mutation, and such an accident might happen as result of consanguinity.[15] In our case there was no relevant family history or other maternal problems.
Recent years have brought about an upsurge in the study of fibroblast-growth-factor receptors, FGFRs, as they pertain to human development. At least fifteen different genetic dysplasias such as Apert syndrome have been linked to FGFRs[16]. The mutation is known to be a Ser252 Trp mutation in the FGFR2 gene[17]. The epidemiology of Apert syndrome is such that the incidence of sporadic births increases exponentially with increasing paternal age, and some investigators have found that this mutation is more common in the sperm of older men[18]. Experiments have indicated that FGFR2 may act as a negative regulator of bone growth.
Conclusion
Until there is a means to correct the molecular defect, we must rely on a strong multidisciplinary approach to patients with Apert syndrome. Neurosurgeons, plastic surgeons, otorhinolaryngologists, orthodontists, pedodontists, ophthalmologists, radiologists, geneticists, pediatricians, and dermatologists must all work in concert to care for patients with Apert syndrome. Early surgical intervention is imperative for optimal outcomes. Subsequent treatment should be tailored to each individual patient's needs.
References
1. Paget J, Lancet 2:1017, 1882 cited in, Human birth defects In: The Developing Human- clinically oriented embryology edited by Moore K L, Persaud TVN ,ed 7th :157, 2003.
2. Shyam Verma, Michelle Draznin: Apert syndrome. Dermatology Online Journal 2005;11 (1): 15.
3. Apert, M. E.: De 1'acrocephalosyndactylie. Bull, et. mem, Soc. med. hop. Paris, 1906;23: 1310-1330.
4. Albuquerque MA, Cavalcanti MG. Computed tomography assessment of Apert syndrome. Pesqui Odontol Bras. 2004 Jan-Mar;18(1):35-9. Epub 2004 Jul 20.
5. Sohi BK, Sohi AS. Apert's syndrome. Indian J Dermatol Venereol Leprol 1980;46:169-72.
6. Harper JI. Genetics and genodermatoses. In: Champion RH, Burton JL, Burns D, Breathnach SM, editors. Rook/Wilkinson/Ebling Textbook of dermatology. 6th ed. Oxford: Blackwell Science; 1998: 425-6.
7. Henderson CA, Knaggs H, Clark A, Highet AS, Cunliffe WJ. Apert's syndrome and androgen receptor staining of the basal cells of sebaceous glands. Br J Dermatol 1995;132:139-43.
8. Batra P, Duggal R, Hariparkash. Dentofacial characteristics in Apert syndrome- a case report. JISPPD;Sep 2002;20(3):118-23.
9. Weese Jl, West WH, Herberman RB, Payne SM, Siwarski JW, Trocotte JG. High affinity and T cell rosette. The effect of clinical manipulation and potential prognostic significance. Am J Sur Oncol 1980;13:145-153.
10. Anil S, Rajendran R, Hari S, Vijayakumar T. Acrocephalosyndactyly (Apert’s syndrome)- a case report. JIDA 1992 Jan;63(1):17-20.
11. Awasthy N, Gupta H. Apert Syndrome. Pediatric Oncall [serial online] 2005 [cited 2005 June 1];2.
12. Blank, C. E.: Apert's syndrome (a type of acrocephalosyndactyly): Observations on a British series of 39 cases. Ann. Human Genet., 24: 151-164, 1960
13. Cooper, H.: Acrocephalosyndactyly with report of a case. Brit.. J. Radiol., 1953;26: 533-538.
14. Vijayalakshmi AM, Menon A. Apert syndrome. Indian Pediatr. 2002 Sep;39(9):876-8.
15. Book JA, Hesselwick L. Acrocephalosyndactyly. Acta Pediatr Scan. 1953;42:359-364.
16. McIntosh I, Bellus GA, Jab EW. The pleiotropic effects of fibroblast growth factor receptors in mammalian development. Cell Struct Funct. 2000 Apr;25(2):85-96.
17. Doutetien C, Laleye A, Tchabi S, Biaou O, Lawani R, Deguenon J, Darboux R, Gnamey D, Bassabi SK. [Apert's syndrome: a case report] J Fr Ophtalmol. 2003 Sep;26(7):738-42.
18. Glaser RL, Broman KW, Schulman RL, Eskenazi B, Wyrobek AJ, Jabs EW. The paternal-age effect in Apert syndrome is due, in part, to the increased frequency of mutations in sperm. Am J Hum Genet. 2003 Oct;73(4):939-47. |
|
|
|
|
|
|